
Modelling the Interactions Between SARS-CoV-2 Spike Protein and Monoclonal Antibodies to Inform Translational Approaches to Treat COVID-19 Infection

Robert Naughton1
1Department of Life Sciences, NUI Galway
Youth STEM Matters, no. 2 (2022); https://doi.org/10.51892/ysm.2.202202R. Naughton, "Modelling the Interactions Between SARS-CoV-2 Spike Protein and Monoclonal Antibodies to Inform Translational Approaches to Treat COVID-19 Infection" Youth STEM Matters, no. 2, 2022.
If you cite this article or use it in any academic activities please let us know by e-mailing us at editor@youthstem2030.org.
Abstract
Introduction
COVID-19
Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) is a positive-sense RNA virus that causes the disease COVID-19 and is responsible for the current pandemic, which has resulted in over 240,000 infections and 4000+ deaths in the Republic of Ireland alone [1]. It is of the family Coronviridae in the subgenus Sarbecovirus, and is an enveloped sphere containing genetic material with trimer spike protrusions on the outside, giving it a crown-like resemblance (corona meaning crown in Latin) [2]. Antigenic drift is an accumulation of genetic variation exhibited by viruses after some time and has been documented in several coronavirus family members, so it is not astonishing that several variants of concern have developed over the past year [3]. The current virus shares 76% identity with SARS-CoV-1, a virus that made headlines in 2003 where it infected over 8000 people, mostly in Asia, and killed over 700 of them. The lower infectivity of the virus and implementation of public health policies like effective quarantining resulted in the virus coming under control one year later [4, 5]. Surprisingly, they both have considerably different spike proteins resulting in a limited number of cross-reactive antibodies [6, 7]. The high diversity of SARS-CoV-2 circulating in various animal reservoirs, such as bats, means that the likelihood of another zoonotic pandemic is likely in the future. Therefore, there is a focus on developing monoclonal antibodies (mAbs) with high neutralisation potential across all family members and novel variants by targeting conserved epitopes on their respective spike proteins to inhibit interaction with the Angiotensin-converting enzyme 2 receptor (ACE2) [6-9].
ACE2
ACE2 is a cellular receptor found in especially high concentrations in the epithelial tissue of the organs of mammals such as the heart, liver, kidney, and lung. Its function is the maturation of the peptide hormone angiotensin used to control blood pressure and vasoconstriction. Decreased expression of ACE2 is associated with cardiovascular diseases, and its mutation causes Hartnup disorder. ACE2 is also the receptor targeted by SARS-CoV, Middle East Respiratory Syndrome (MERS) and SARS-CoV-2 [10]. SARS-CoV and SARS-CoV-2 have >75% identity. The spike protein (S protein) binds to the receptor where it is endocytosed or directly fused with the plasma membrane and once inside hijacks the cell machinery for viral production (Fig. 1) [4]. As a result, there is considerable interest in understanding the interaction between the viral spike and ACE2 binding sites. SARS-CoV-2 spike protein exhibits a dynamic swinging motion on binding, which is thought to contribute to its higher infectivity [4]. Variants like B.1.1.7 (Alpha) have notable mutations such as N501Y which is believed to result in the increased infectivity of the strain; estimated to be up to 90% more transmissible [11]. Therefore, there is a clear need to understand the interaction between the S protein and ACE2 as it is the way which the virus infects the host. With an inability to downregulate or block the receptor without causing harm to an individual, there is a necessity to target the spike protein by developing mAbs to block its binding to ACE2 [10, 12].

Figure 1 - SARS-CoV-2 entry mechanism via receptor-mediated endocytosis using ACE2. The full spike trimer on the virus binds to an ACE2 receptor on the cell's surface. The TMPRSS2 protease cleaves part of the spike protein allowing for the activation of the S2 domain. This allows for the fusion of the viral and cell membranes allowing for the entry of the viral RNA. The RNA is then copied by host cell machinery, creating more viral particles [4, 10, 12]. Figure constructed by the Youth STEM Matters Publication Team, advised by Author.
Spike Protein
Spike protein or S protein is a homotrimer glycosylated protein on the surface of the SARS-CoV-2 virus and is responsible for its viral entry [13]. This homotrimer comprises of three identical units each with two subunits known as S1 and S2 [13]. The S1 is made up of the N Terminal Domain (NTD) and C Terminal Domain (CTD), with the former containing the receptor-binding domain (RBD), which binds to the ACE2 [10, 13]. The S2 subunit is activated upon binding of the S1 to host ACE2, proteases then cleave part of S2, aiding in viral entry (Fig. 1) [10]. The RBD of the S1 directly interacts with ACE2, N501Y mutation in the Alpha variant is in the RBD and results in its higher infectivity [14]. Mutations, such as E484K and K417N/T present in both B.1.351 (Beta) and P.1 (Gamma), are also on the RBD, affecting antibody recognition, however, these do not directly modify infectivity [3]. The spike protein, specifically the RBD of the S1 subunit, is of high mutation due to its key function in the infection of host cells [3]. Selective pressure causes antigenic drift, resulting in increased infectious strains and escapes mutants thanks to Darwinian evolution [3, 8]. The spike protein's structure, like all proteins on a molecular level, confers functionality. As a result, efficient study, development, and computer model testing can show potential mutation sites as novel epitopes for mAbs to prevent widespread reinfection [12, 15].
Receptor Binding Domain
The Receptor Binding Domain (RBD) is an area of much significance found on the NTD of the S1 part of the spike protein and it binds to the peptidase domain of the ACE2 receptor, specifically at nucleotides 319-541 [10]. The Receptor Binding Motif (RBM) is a small region found within the RBD at nucleotides 438-506, this motif directly interacts with the ACE2 (Fig. 2) [13]. The spike protein's homotrimer configuration means it has three RBDs in total that can individually be in up or down confirmations granting it open or closed forms [13]. Different mAbs have different preferences on the confirmations of the RBDs to bind effectively, but the vast majority can only attach to an RBD in the up confirmation. Structural analysis of the spike trimer has shown its dynamic nature and a swinging motion can be seen when the RBD binds to the ACE2 receptor, which could explain its increased infectivity compared to related viruses [4].

Figure 2 - SARS-CoV-2 spike protein-coding domains. Spike protein has several different domains pictured above as different colours. These various domains each have their specific function, but much of our interest lies in the RBD, which contains the RBM. Other domains include NTD, N-terminal domain; SD1, subdomain 1; SD2, subdomain 2; FP, fusion peptide; HR1, heptad repeat 1; HR2, heptad repeat 2; TM, transmembrane region; and the IC, intracellular domain [16]. Figure constructed by the Youth STEM Matters Publication Team, advised by Author.
Variation and Conservation
B.1.1.7, P.1 and B.1.351 are the variants that have caused concern across the globe due to their mutations, yet conservation remains high (Fig. 3). Each has several mutations but ones found on the RBD, such as K417N, E484K, and N501Y, have resulted in the proposed antibody evasion and increased infectivity [17]. Notably, both natural and artificial antibodies have epitopes mostly found on the RBD of the spike protein. These are the most effective as they sterically block the binding of the virus to the ACE2 [13]. Through Antibody Dependant Enhancement (ADE), whereby ineffective antibodies increase the entry of virus into the host cells. Therefore, it is a fear when developing treatments and vaccines for a pathogen and has been found to occur with SARS-CoV and MERS; albeit there is no current evidence that this is a problem with SARS-CoV-2 [13, 18]. The variation seen in the RBD and the bias for antibody targeting of that region, there is evidence of escape mutations occurring. There is proof of specific immune memory in the form of IgG antibodies in individuals who suffered a mild infection for up to three months post-infection [9]. Unfortunately, due to the continuing development of the pandemic, it is not currently known precisely how long individuals are immune and if this will affect the efficacy of the vaccines in the long run [19].
Monoclonal Antibodies
mAbs are small Y-shaped proteins produced by plasma cells. Monoclonal refers to the fact they are copies of each other. mAbs are highly varied and function to bind to specific antigens. Antibodies are naturally produced by the host's immune system and are usually selected to bind efficiently to the antigen of interest [21]. Antibodies neutralise the virus by binding to the SARS-CoV-2 S protein, often sterically hindering future binding to ACE2 receptors (Fig. 4) [12, 16]. Since 2015, mAbs have maintained their position as the highest-selling pharmaceutical drug [22]. Antibodies are developed in response to an antigen therefore, people naturally infected by the virus or inoculated produce them in high concentrations in response to future contact with the virus [12]. Antibodies can also be administered intravenously in the form of antibody cocktails to severely ill individuals who do not have enough time to produce them in large enough quantities [23]. Most of the epitopes targeted by monoclonal antibodies are found in the RBD of the spike protein [12]. Variants such as P.1 and B.1.351 have mutations like E484K or K417N; single mutation variants created in the lab have shown that the neutralisation of certain mAbs was reduced up to ten-fold by them [8].
Selective pressure has resulted in the natural creation of these variants. It was hypothesised that P.1 was responsible for the high rate of infection in Manaus, Brazil, where 75% population was thought to have a high seroprevalence of antibodies targeting the principal variant of SARS-CoV-2 [24]. Nevertheless, there was a considerable increase in hospitalisations in January 2021 for the disease, pointing to a lack of protection from antibodies against novel strains such as P.1, even when antibodies were shown to be at high levels three months post-infection [19, 23]. Though this study was somewhat flawed with a limited amount of data and extrapolation to estimate infections, it did show the virus's mutagenic potential [24]. Antibody cocktails targeting separate epitopes have been shown to prevent the formation of escape mutants though they are only available for sick patients, and prevention is better than treatment [12]. Mapping of potential mutations and artificial intelligence to predict novel mAbs for development are ways to get ahead of the virus’s antigenic drift; these programs have shown great accuracy though they remain highly complex [15, 24].
mAb Therapies
In October 2020, former US President Donald Trump had contracted COVID-19; due to his age of 73 and high BMI, he was defined as being high-risk. He was inoculated with the experimental Regeneron mAb cocktail (REGN-COV-2), and quickly recovered, illustrating the benefits of such treatments [26]. mAb cocktails are therapeutic mixtures of two or more mAbs to significantly affect the neutralisation of an antigen, in this case, SARS-CoV-2 spike protein. The selective pressure on the virus from antibodies has created several variants that have been shown to possess mutations reducing the neutralisation effectiveness of some antibodies tenfold [8, 17]. Studies show that antibody cocktails containing mAbs that effectively target separate epitopes have great potential in treating disease and preventing escape mutants. The statistical probability of a virus possessing mutations at both epitopes is highly improbable, reducing the risk of escape mutants [8, 22]. Artificial intelligence in the form of neural networks continues to generate high throughput approaches to drug discovery. Scientists have developed a method to map all the antibody escaping mutations on the RBD of SAR-CoV-2, applying it to ten different antibodies [15]. Mapping, next-generation sequencing, and creating phage display libraries have allowed for the quick and accurate selection of highly effective mAbs for use in cocktail treatments, as seen by the P17 and H014 cocktail in the results [22, 26]. They are rated based on neutralisation capability, area of binding, and compatibility with other mAbs [15].

Figure 3 - SARS-CoV-2 spike protein showing the conservation between the three variants: B.1.1.7, B.1.351, P.1 and WT virus. Complete conservation is illustrated by red, single mutations that are not present in any other variants appear white, and residues mutated in more than one variant are blue. (A) Bottom view of the S protein. (B) Side view of S protein. (C) Top-down view of the S protein. PDB entry 6VXX [20].
Nanobodies
Antibodies are traditionally Y-shaped proteins with the effective binding region at the tip of the Y shape. Nanobodies (Nbs) are naturally produced by members of the camelid family and are 10% the size of a normal antibody while still having the same specificity and affinity (Fig. 4) [27]. They also are potent inhibitors of viruses, and their small size allows easier binding [28]. An experimental anti-SARS-CoV-2 nanobody isolated from Vicugna pacos (Alpaca) called Ty1 has shown great potential. Usually, the S protein presents itself with one RBD up and two down. Unlike other mAbs, Ty1 can bind to all three RBDs regardless of their configuration [28].
Further studies in this area created heterobivalent Nb that bond to Fragment Crystallisable (Fc) stems, which are the tails of the antibodies. This results in a ten-fold increase in neutralisation. This improvement is due to the two different epitopes the Nbs can bind, while the Fc stem gives them a more significant steric hindrance in blocking the binding between RBD and ACE2 [29]. Testing the B.1.1.7 variant again showed that even with the improved bonding between RBD and ACE2 via N501Y, the Nbs were still highly successful in impeding their interaction. However, research needs to be done on P.1 and B.1.351, which have mutations that directly affect mAb binding [29]. There is a massive potential to produce Nbs in bacteria as antibody cocktails for the treatment of COVID-19 due to their cheap production, ease of manipulation, scalability, high stability, and excellent neutralisation to prevent the selection of escape mutants [27, 28].

Figure 4 - Neutralisation of SARS-CoV-2 by mAbs, mAb cocktails, and nanobodies. These three types of treatment of an active form of a COVID-19 infection have their own advantages; however, they all work similarly. They bind themselves to the S protein of the virus, sterically hindering binding to the ACE2 receptor, thereby preventing viral entry and rendering the viral particle neutralised. Figure constructed by the Youth STEM Matters Publication Team, advised by Author.
Aims
Firstly, this paper aims to produce a satisfactory structure analysis of the interactions between the wild type of SARS-CoV-2 S protein and the ACE2 receptor/mAbs using Chimera. Secondly, mutating the highlighted key residues and visualising points of contact between these molecules would allow the understanding of how the mutations in the variants confer increased infectivity or mAb resistance. By choosing mAbs that exhibit huge decreases in neutralisation against the variants, it would determine what intermolecular forces the E484K and K417N/T mutations change. Thirdly, imaging the interactions between the variant SARS-CoV-2 S proteins against proposed mAbs, and Nbs would aid in the development of novel treatments and vaccines.
Methods
Sequence Selection
The variant SARS-CoV-2 spike proteins of B.1.1.7, P.1, and B.1.351 were analysed, compared, and contrasted against the WT Wuhan variant, using only four primary sequences (Appendix). Since the viruses mutate constantly, and there are often differences seen between patients infected with the same variant, it was challenging to get reference sequences. As a group, we researched the mutations seen in each variant and supplied the data to Dr. Kevin Sullivan. He derived the WT spike sequence from NCBI with the accession number YP_009724390.1. From there, Dr. Sullivan manually put together the three variant sequences using the data on the PANGO lineages database [30]. Finally, he was able to create reference sequences for the four variant spike proteins that would then use for alignment and conservation visualisation (Table 1).
Table 1 - Variants of concern, amino acid mutations, the mutations of interest and how they affect interactions between the virus and ACE2/mAbs. Each variant has many different mutations; though most have little to no known effect on the virus, some confer increased infectivity or mAb evasion.

Protein Selection
All proteins were selected from literature via protein database (PDB) by the RCSB (RCSB PDB: Search) [31]. PDB has a page dedicated to SARS-CoV-2 proteins, divided into the different proteins of each type, spike proteins being of the most interest. Hundreds of structures are available, from complete spike proteins to RBDs, bound and unbound, and interactions between ACE2 and mAbs. PDB proteins have unique four-character reference names that can be copied and loaded straight into the visualisation software Chimera (Table 2).
Table 2 - The PDB file entries, descriptions, and their use in this project. Over 15 structures were analysed in this project, but only the most relevant data was kept for the project results.

Multiple Sequence Alignment
Alignment of the multiple sequences was performed using Seaview with the CLUSTAL OMEGA algorithm (PRABI-Doua: SeaView) [32]. The four sequences of the three variants and wild-type SARS-CoV-2 spike proteins were supplied and edited by Dr. Kevin Sullivan. Upon loading into the program, they were aligned using CLUSTAL Omega. Once aligned, the file was saved as a PDF for visual inspection as well as a .aln file for use in conservation analysis using Chimera X and Web Logos (Appendix).
Protein Visualisation
All protein visualisation in results was undertaken using the freeware program "Chimera" created by UCSF [33]. Chimera is a protein visualisation tool that allows for the imaging, manipulation, and analysis of protein structures on the atomic and molecular scale. Chimera has many different tools available. A significant use of Chimera for this project was viewing the interactions between the spike proteins and ACE2 or mAbs; it was used to view individual bonds between the molecules and mutate single residues to simulate the different interactions seen between the variants and ACE2/mAbs.
PDB entries from Table 2 were used in this project of interest and loaded into Chimera by using the fetch function. Enabling the command line was highly useful for the selection of the residues of interest, as various commands can be found on the Chimera website. Mutation of single residues was done using the rotamers function in structure editing for the most accurate representation of mutations. Contacts between residues were illustrated with the contacts/clashes tool in structure analysis. Production of high-quality images was done using the "publication 3" pre-set and saving images as transparent PNG files. Residues, molecules, and other parts of the proteins were labelled with specific colours depending on what they represented (Table 3). Chimera X was used in the generation of full spike high-definition renders as well as video renders in the presentation created (Fig. 5). Chimera X was also used to generate a conservation map of the spike protein (Fig. 3) [20].
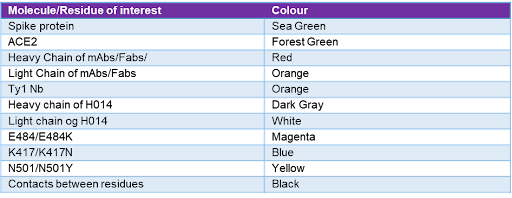
Table 3 - Molecule or residue of interest and their corresponding colourations in the Chimera images. Each residue or molecule of interest has a corresponding colour, as seen in the visualisations of the results section. This allows for a simpler understanding of the results.
Results
Structural and Mechanistic Analysis of SARS-CoV-2 Spike Protein Interactions

Figure 5 - SARS-CoV-2 Spike protein with 3 RBDs down. (A) Side view of S protein trimer, which is formed out of three identical monomers, each seen as a different colour. (B) The top-down view showcases the RBDs in down conformation, creating a triangular pin-shaped structure. PDB entry 7BNM [20].
The RBDs are found at the top of the S protein. These contain the RBM, which directly interacts with the ACE2 receptor and initiates endocytosis, thereby allowing viral replication to occur (Fig. 5). The spike is glycosylated and covered in oligosaccharides, but that cannot be easily viewed in Chimera.
SARS-CoV-2 binding to ACE2
Each of the monomers has an RBD that can bind a single ACE2 molecule (Fig. 6). Critical residues for binding on the RBD are Q493, N501 and N481-487; key mutations in all three variants are on one or more of these residues [4]. 94% of unbound trimmers that float around are in the tightly closed state, which helps them avoid detection and binding by antibodies that specifically bind to the RBD; in MERS and SARS-CoV, this is less than 50% of the time. The 22 N-linked glycans found on the spike protein, but not pictured here, are also thought to aid in the prevention of detection by the human immune system [4]. The RBD is only 194 residues in length, yet without it, the spike protein would be ineffective [16].

Figure 6 - ACE2 bound to SARS-CoV-2 S protein. (A) Homotrimer spike protein bound to three ACE2 molecules. (B) Single spike monomer bound to one ACE2 molecule. Colouring as in Table 3. PDB entry 6MOJ.
Hydrogen bonds form between crucial residues on the Alpha chain on ACE2 and the RBM of the RBD on the S protein; these interactions allow for the 20x increased binding by the virus compared to SARS-CoV [29]. The loop T470-T484 and residue Y505 are highly important for the recognition of the RBD by ACE2 (Fig. 7) [4]. There is a swinging motion seen when the spike protein comes near the ACE2 receptor. It causes one of the RBDs to switch into the up position where it then binds directly with ACE2; next, fusion occurs, and the viral RNA enters the cell for replication [4]. An essential residue D614 forms four hydrogen bonds/salt bridges with the help of neighbouring residues [4]. The binding of ACE2 to the RBD causes conformational changes in the S1 leading to a reduced interface between itself and S2, eventually allowing for fusion with the membrane [4].

Figure 7 - Interactions between RBD of spike protein and ACE2, with residues that exhibit hydrogen bonding. (A) Interaction with WT spike protein and N501 residue. (B) Interaction with B.1.1.7 variant that has the N501Y mutation. Colouring as in Table 3. PDB entry 6MOJ.
Interactions Between mAbs and WT SARS-CoV-2 Spike Protein
This mAb’s effectiveness is drastically affected by the E484K and K417N mutations found in some variants (Fig. 8) [8]. There is limited H-bonding seen in C and between the residues of interest, though the overall interaction between the mAb and RBD can be seen in D.

Figure 8 - Visualisation of COVOX-40 Fab bound to S protein with focus on the RBD. (A) Spike trimer with one RBD in the up conformation bound to a COVOX-40 Fab. (B) Isolated monomeric spike protein chain bound to COVOX-40 Fab. (C) Contacts between N501, E484 and K417 with COVOX-40. Limited H-bonding between the residues of interest. (D) Contacts between full RBD and COVOX-40. Colouring as in Table 3. PDB entry 7ND3.
SARS-CoV-2 Variants and Their Adaptions
B.1.1.7 and Infectivity
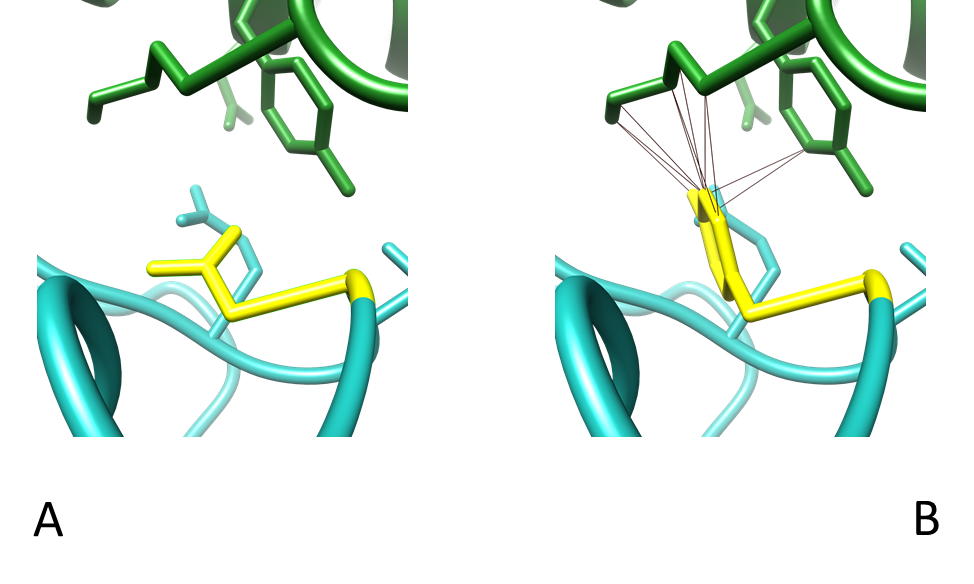
Figure 9 - ACE2 bound to SARS-CoV-2 spike protein interaction focusing on contacts between residue 501 of RBD and ACE2. (A) N501 has no contacts with ACE2. (B) N501Y showing 10 points of contact with residues Y41 and K353 of ACE2. Colouring as in Table 3. PDB entry 6MOJ.
The increased infectivity of the Alpha variant is associated with the mutation N501Y. It is shown that the mutant Y501 interacts with the residues Y42 and K353 on the ACE2 receptor (Fig. 9) [14]. Original N501 has two non-bonded interactions with Y41 and K353 at a 3.5-angstrom distance, whereas Y501 forms a H-bond with K353 and non-bonded interactions with D38, Y41 and K353 at a 3.5-angstrom distance (Fig.9) [14]. Not only that, but more H-bonds were found to be formed between Y501 and residues Y500, Y501, G502, V503 and Y505 on the RBD, further increasing the interaction between it and ACE2 [14].
Chen et al. analysed the WT spike protein ACE2 interaction and found that one of the several critical binding residues is N501. Various mutations had a limited impact on mAb neutralisation in B.1.1.7 compared to B.1.351 and P.1. Loss of neutralisation was minimal and only seen in three mAbs, including COVOX-40, whereas the other 15+ antibodies tested showed no statistically significant difference [34].
P.1/B.1.351 Variants and mAb Escape
K417N is a mutation seen in the B.1.351 variant, and, at the same residue, there is the mutation K417T in P.1 (Fig. 10) [3]. They also found the K417N mutations to be specifically responsible for the loss of neutralisation of four antibodies, while E484K rendered five of the 18 antibodies that tested ineffective [3]. Using single mutant viruses they made in the lab, Diamond et al. found that the K417N mutation was responsible for a ten-fold reduction in the neutralisation of the mAb COVOX-40 and other mAbs. However, all the other mAbs they tested showed no difference [8]. Another group found this same situation to be responsible for the complete loss in the neutralisation of other mAbs like 910.30, and cooperation was seen between K417N and E484K, causing a sharp decrease in efficiency of REGN10933 [17]. Preliminary research has shown that these variants impact some of the approved vaccines in this study, specifically Pfizer-BioNTech (BNT162b2), and, in some cases, reduce neutralisation by four-fold [8]. Due to content restraints, it was not possible to include visualisations of the interactions between K417T and mAbs; however, based on the data in this study and the current literature, the mutation appears to reduce mAb neutralisation similarly to K417N [35].

Figure 10 - Interactions between residue 417 of the RBD and the heavy and light chain of COVOX-40 Fab. (A) Contacts between WT K417 and COVOX-40 which are Y52 and D92. (B) Contacts between K417N mutant with COVOX-40, which are Y52 only. Colouring as in Table 3. PDB entry 7ND3.
E484 has H-bond interactions between itself and mAbs. In contrast, the mutation to K484 destroys this; for example, the mAb CVO5-163 forms H-bonds and a salt bridge with itself and E484. Thus, there are no H-bond interactions in the mutant variants, leading to a vast loss in neutralisation with COVOX-40 (Fig. 11) [3]. E484-F486 is a key hydrophobic area of the RBD used by many mAbs, having extensive interactions with them, and is accessible from several angles of attack [36].

Figure 11 - Interactions between the residue 484 of the RBD and its surrounding residues on the same chain. (A) Contacts between WT E484 and nearby residues I472, C488 and F490 on the RBD. (B) Contacts between E484K mutant with residues I472 and C488 on the RBD. Colouring as in Table 3. PDB entry 7ND3.
Combative Therapies Against the Variants
Single mAbs that Target Conserved Epitopes
With a spillover event attributed to the first human infection of SARS-CoV-2 and the increase of intensive animal agriculture practices putting society in more contact with them, there is a need to prepare effective treatments against upcoming zoonosis events [7].
ADG-2 is a mAb discovered by Rappazzo et al. that effectively neutralises over 14 sarbecoviruses found in humans, bats, and pangolins [7]. Structural analysis showed that its unique angle of approach and its ability to recognise a conserved domain on the spike protein are the reasons for its effectiveness (Fig. 12) [7].
A homologous Fab, 6APC, docked into a low-res EM density map of ADG-2, was kindly supplied by Dr. Jason McLellan, UT Austin. This allowed the interpretation of ADG-2's angle of binding but didn't allow for the analysis of the individual residue interactions (Fig. 12).

Figure 12 - Detailed visualisation of the angle of binding by pseudo-ADG-2. (A) RBD and pseudo-ADG-2 structure illustrating ADG-2's unique angle of approach binding epitope. (B) Closer look describing the proximity of N501 to pseudo-ADG-2 structure in comparison to E484 and K417. Colouring as in Table 3. PDB file supplied by Daniel P Wrapp.
mAb Cocktails
Analysis of the human antibody library enabled the finding of compatible antibodies that would synergise, not compete with each other by binding to unique epitopes of the S protein, preventing the selection of escape mutants [22, 26]. This technique resulted in the development of an antibody cocktail containing H014 and P17, both of which have high neutralisation, with the former binding to a conserved epitope (Fig. 13) [27]. HIV selective mutations have occurred in the past to antiviral therapies, and one such way of combating this was the development of mAb cocktails [27].

Figure 13 - P17 and H014 antibody cocktail bound to all 3 S protein homotrimers focusing on the S protein RBD. (A) P17 and H014 antibody cocktail attached to S protein homotrimer. (B) Top-down view of S protein homotrimer bound to the two different antibodies. (C) Detailed visualisation of binding of P17 and H014 to the RBD with residues of interest highlighted. Colouring as in Table 3. PDB entry 7CWN.
All three of the RBDs are in the up position here because they are open. The antibodies bind distinct epitopes on the viral protein and do not overlap, which is vital in preventing mutagenic escape. The RBD and H014 exhibit strong bonding via a unique epitope (Fig. 14).

Figure 14 - Illustration of H014's unique binding epitope and the contacts which allow for this. (A) Contacts between RBD and H014. (B) Another view of the contacts between the RBD and H014. Colouring as in Table 3. PDB entry 7CWN.
There is some interaction between P17 and E484 (Fig. 15). There is a vast improvement in neutralisation and binding when these H014 and P17 are used together instead of separately, giving them a synergistic nature. P17 binds to the top of the RBD, thus blocking ACE2's binding site. H014 attaches to the RBD side and creates an exterior layer while P17 forms a cap layer; together, they act as a shield [27].

Figure 15 - Interactions between P17 and S protein RBD with a focus on RBD residue 484. (A) Hydrogen bonds formed between WT E484 of RBD and P17. (B) Hydrogen bonds formed between mutant E484K and P17. (C) Contacts shown between WT E484 and P17 at residues H35, R96 and T101. (D) Contacts shown between mutant E484K and P17 at R96 only. Colouring as in Table 3. PDB entry 7CWN.
Humanised Camelid Nanobodies
When mutations arise, antibodies can lose their ability to bind to the epitope; they even caused ADE in SARS and MERS animal models [18]. Nanobody research into effective SARS-CoV-2 treatments has been highly productive. These tiny antibodies confer high specificity effectively while remaining cheap and stable, all the while being easily manipulated [27, 28]. Their small size can be a limiting factor since the effectivity of mAbs against SARS-CoV-2 has been associated with their steric hindrance preventing the binding of the S protein to the receptor [13]. Based on this, Ma et al. created heterobivalent antibodies from nanobodies, increasing the MW by six-fold; these had a much higher affinity than mono Nbs, even more so than patient's antibodies [29]. Only one of the seven antibodies identified could not bind successfully to prevent the S protein from binding to ACE2; however, bonding it to an IgG1 Fc stem improved its neutralisation by over 70x [27, 28]. Alpaca VHH (an antibody fragment) shares a high degree of homology with VH3 in humans, therefore there should be low immunogenicity for patients' treatment, reducing the risk of allergic reactions. Genetic modification and affinity maturation could be used to improve antiviral function, allowing for treatment [29]. Testing the Alpha variant has already shown that, even with the increased binding due to N501Y, nanobodies are successful at blocking the interactions between the RBD and ACE2 [29]. Ty1 is one such example of an Alpaca derived anti-SARS-CoV-2 nanobody found by another research group (Fig. 16). Ty1 exhibits the unusual ability to bind to both closed and open conformations of the RBD, whereas most mAbs can only attach to the open form. With the vast majority of free-floating spike proteins shown to be in closed confirmation to avoid immune detection, Ty1 could effectively aid in virus suppression [4, 28].
The nanobodies bind to all three RBDs, sterically hindering the interaction between the S protein and the ACE2 effectively (Fig. 16). Ty1, like other Nbs, is 10% the size of other mAbs but still provides similar, if not better, neutralisation [28]. Hanke et al. effectively cloned and expressed Ty1 in high quantities in bacterial cultures, making potential production cheap and accessible [28].

Figure 16 - Visualisation of S protein bound to three Ty1 Nbs, with a focus on RBD interactions. (A) Full spike homotrimer bound to three Ty1 Nbs, with 2 RBDs in up conformation and one in down. (B) Isolated monomeric spike protein chain bound to Ty1 with RBD in up conformation. (C) Detailed imaging of the interactions between RBD and Ty1 with residues of interest highlighted. Colouring as in Table 3. PDB entry 6ZXN.
There seems to be no such relationship between Ty1 and N417, so there should be little change in its binding with variants possessing the N417K/T mutation (Fig. 17). The literature details the interactions between Ty1's complementary determining region (CDR) domains and the RBS. These include T470, Y449 and, worryingly, V483-E484 [28].

Figure 17 - Illustrated contacts between Ty1 and 484 of RBD as well as the full RBD of the S protein. (A) Contacts between WT E484 and Ty1 at residues 33-35. (B) No contacts found between mutant E484K and Ty1. (C) Complete visualisation of contacts between Ty1 and RBD. (D) Hydrogen bonds between Ty1 and RBD. Colouring as in Table 3. PDB entry 6ZXN.
Discussion
Structural and Mechanistic Analysis of SARS-CoV-2 Spike Protein Interactions
SARS-CoV-2 Binding to ACE2
Extremely high level of binding exhibited by the contacts seen between the S protein RBD and ACE2 (Fig. 6, Fig. 7). It is highly interesting that the seemingly minor change of a single amino acid substitution causes the drastic increase in affinity between the RBD and ACE2, translating to higher infectivity of the B.1.1.7 variant [14].
Interactions Between mAbs and WT SARS-CoV-2 Spike Protein
Analysis of the many structures available on PDB showed a clear bias for mAbs targeting the RBD of the SARS-CoV-2 spike protein. In nearly all the structures analysed, there were precise interactions between that part of the spike protein; analysis showed that a lot of their high neutralisation was due to the fact they were binding to the RBD, thus sterically blocking the interaction between the spike and ACE2 receptor. There may have been many more mAbs that bind to epitopes and are not on the RBD, but their unattractively low efficacy meant they were most likely filtered out during mAb discovery.
There undoubtedly is tight binding between COVOX-40 and RBD, as seen by the high number of contacts, close packing and proximity between the two with the surface model (Fig. 8).
SARS-CoV-2 Variants and Their Adaptions
B.1.1.7 and Infectivity
Y501 can be seen forming increased contacts with residues on the ACE2 receptor , especially to the residues of interest on ACE2 Y42 and K353 on ACE2, increasing the binding (Fig. 9). This provides further evidence as to why this mutation makes such a difference in binding [4].
P.1/B.1.351 Variants and mAb Escape
The mutation K417N drastically increases the space between the residues of the RBD and COVOX-40, which likely aids in reducing the contacts it has with neighbouring residues (Fig. 11). Lysine is positively charged while Asparagine is polarly uncharged; therefore, salt bridges could form between WT RBD and COVOX-40. Mutation to Asparagine would not allow for this salt bridge and may be responsible for the reduced neutralisation caused by reduced binding exhibited in K417N mutants [34].
Residues N481-487 are key binding sites, hence why a mutation at 484 has such an effect on neutralisation [4]. The interaction of three different spike protein structures with various mAbs was tested, but their E484 can be seen pointing in the opposite direction and not directly interacting with the residues of the mAbs (Fig. 8); this is also suggested by several papers [4, 7, 8, 22]. Phe486 of the RBD directly interacts with the mAbs in the structures, so perhaps E484 stabilises this interaction and acts as a supporting unit for these neighbouring residues that directly interact with the mAb. If that is the case, it is interesting that there have not been any widely recorded mutations of these neighbouring residues causing reduced mAb efficacy. The mutation to Asparagine breaks the bond between itself and D92 (Fig. 10), and this change strangely affects the binding of the mAb when there is such a vast array of binding epitopes (Fig. 8D).
Combative Therapies Against the Variants
Single mAbs that Target Conserved Epitopes
Although the simulation of the interactions between ADG-2 and the S protein was only tested on B.1.1.7 as it was the only variant available at the time, its unique epitope of recognition being D405 and effectiveness across a broad group of coronaviruses would suggest that it is also effective against B.1.351 and P.1, though further research should be done to confirm this [7]. ADG-2 and its study could lead to novel mAb therapies and effective broad-spectrum vaccines [7]. The conserved epitope it binds may also help prevent mutant variants that develop under selective pressure as D405 may be essential to the spike structure (Fig. 12) [6]. Its effectiveness would help prevent ADE, which has been seen in clinical trials for vaccines for MERS and SARS-CoV; however, there is limited evidence that SARS-CoV-2 causes ADE [18].
mAbs Cocktails and Booster Shots
H014 interacts with the residues 300-400 of the RBD that does not have many mutations, so the probability of an effective mutant variant that can escape is improbable (Fig. 14). While P17 recognises a conformational epitope on the RBM and can bind both open and closed RBDs, one of the 14 essential epitopes it has with the RBD is E484 (Fig. 15). This may cause issues with variants, specifically P.1 and B.1.351, and so it should be researched [27]. As a result of these variations, booster vaccines may be required in the future to reduce the chance of escape by novel variants which, if not monitored, could drastically reduce the protection exhibited by vaccinated populations against the virus [37].
H014 targets an area at the side of RBD which is not in proximity to the three residues of interest, suggesting that its efficacy would not be affected outright by the variants (Fig. 14). However, due to the area it binds, it is less effective than mAbs that target the top of RBD, hence why it is utilised in a cocktail with P17.
Humanised Camelid Nanobodies
Analysis of the interactions between the RBD and a Ty1 molecule shows that there is most likely an interaction between E484 and the Nb (Fig. 16), raising the question of what effect the E484K mutation has on this relationship. Based on the other examples of antibodies, E484K reduces the neutralisation capability of antibodies; coupled with the tiny size of the nanobody, the effects could be highly reductive.
There seems to be no such relationship between Ty1 and N417 (Fig. 17), so, mutation or not, there should be little change to its binding with variants possessing the N417K/T mutation. The literature details the interactions between Ty1's CDR domains and the RBS; these include T470, Y449, and, worryingly, V483-E484 [22]. Research should be done on testing the Nb with variants to ensure it is still highly effective before more resources are put into this specific Nb [28].
Conclusions and Future Perspectives
There is hope that the rollout of vaccines will reduce cases and outbreaks sufficiently enough to bring life back to some form of normality. However, with the advent of the variants like P.1, B.1.1.7 and B.1.351, there is proof that their increased infectivity and mAb avoidance may make this a more complicated transition. From the analysis of ACE2, the SARS-CoV-2 spike protein of three variants and wild type, mAbs, cocktails and nanobodies, a detailed understanding of the interactions was via Chimera. The mutations exhibited by the variants are challenging. However, with the advent of improved mAb cocktails, mAbs targeting conserved epitopes, and nanobodies for widescale production, the arms race wages on.
Research on the ADG-2 mAb that targets a conserved epitope on the sarbecovirus, makes it effective against at least 15 members, and an exciting drug for clinical trials. Due to the high mutation rate of coronaviruses and their ubiquity in bat populations, with one study finding over 150 different coronaviruses in a Chinese cave, there is the potential to stockpile ADG-2 as a treatment for the next sarbecovirus outbreak [7, 38]. mAb cocktails have detailed the benefits of using two different mAbs simultaneously for increased neutralisation and reduction in escape mutants, while requiring fewer resources for development than creating novel mAbs and other treatments [13, 22, 26]. Nanobodies have shown that they are highly producible, cheap, stable and effective in animal trials as anti-SARS-CoV-2 treatments. However, they must be tested in humans with Fc stem attachments to prove their effectiveness as their different nature may result in less effective results in human subjects [26, 27].
Acknowledgements
I would like to thank Dr Kevin Sullivan at the National University of Ireland, Galway for his supervision and mentoring throughout this assignment. His expertise and scientific knowledge were essential from choosing the title to the final draft. I would also like to thank Martti Tolvanen and Csaba Ortutay for their organisation and running of the two weeks bioinformatics course. This teaching in this crash course was essential to my project and I used nearly all the techniques taught to me in the creation of this project. Huge thanks to Daniel P Wrapp of Dr. Jason McLellan's laboratory for sending me the relevant data on ADG-2, without it I would not have been able to produce my own figures. Much appreciation for Orla Mulcahy who spent time reading through my work and suggesting edits. I would also like to thank my fellow classmates Fiona Prendergast, Eimear O'Brien, Cian Parsons and Mark Lawler for sharing their results, methods and advice on this area. Much appreciation to Onika Tanya Maraj and Amala Ratna Zandile Dlamini, their art inspired me to keep working at this project and persevere when obstacles were encountered. Many thanks to the references below for providing me with all this information for free. Huge gratitude to the team at Youth STEM 2030 for doing multiple reviews of my paper, publishing it and supporting young people in STEM across the world. I would finally like to thank my parents Paul and Patricia Naughton, they taught me to work hard and have always supported my interest in science from preschool to university..
Author's Notes
All figures, or tables, were created by the Author, unless otherwise mentioned in the description provided of said figure.
References
[1] E. Dong, H. Du, and L. Gardner, “An interactive web-based dashboard to track COVID-19 in real time,” Lancet Infect. Diseases, vol. 20, no. 5, pp. 533–534, 2020. Available: https://doi.org/10.1016/S1473-3099(20)30120-1.
[2] G. Forni and A. Mantovani, , “COVID-19 vaccines: where we stand and challenges ahead,” Cell Death & Differentiation, vol. 28, pp. 626-639 , 2021. Available: https://doi.org/ 10.1038/s41418-020-00720-9.
[3] M. Yuan et al., “Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants,” bioRxiv, 2021. Available: https://doi.org/10.1101/2021.02.16.430500.
[4] C. Xu et al., “Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM,” Science Advises, vol. 7, no. 1, 2021. Available: https://doi.org/10.1126/sciadv.abe5575.
[5] S. K. Chew, “SARS: how a global epidemic was stopped,” BulletinWorld Health Organisation., vol. 85, no. 4, p. 324, 2007. Available: http://dx.doi.org/10.2471/BLT.07.032763.
[6] M. Yuan et al., ‘A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV’, Science, vol. 368, no. 6491, pp. 630-633, 2020. Available: https://doi.org/10.1126/science.abb7269.
[7] C. G. Rappazzo et al., “Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody,” Science, vol. 371, no. 6531, pp. 823–829, 2021. Available https://doi.org/10.1126/science.abf4830.
[8] M. Diamond et al., “SARS-CoV-2 variants show resistance to neutralization by many monoclonal and serum-derived polyclonal antibodies,” Research Square, 2021. Available: https://doi.org/10.21203/rs.3.rs-228079/v1.
[9] S. H. Hodgson, K. Mansatta, G. Mallett, V. Harris, K. R. W. Emary, and A. J. Pollard, “What defines an efficacious COVID-19 vaccine? A review of the challenges assessing the clinical efficacy of vaccines against SARS-CoV-2,” Lancet Infect. Diseases, vol. 21, no. 2, pp. 26–35, 2021. Available: https://doi.org/10.1016/S1473-3099(20)30773-8.
[10] R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo, and Q. Zhou, “Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2,” Science, vol. 367, no. 6485, pp. 1444-1448, 2020. Available: https://doi.org/10.1126/science.abb2762.
[11] N. G. Davies et al., “Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England,” Science, vol. 372, no.6538, 2021. Available: https://doi.org/10.1126/science.abg3055.
[12] Y. Sun, B. Kobe, and J. Qi, “Targeting multiple epitopes on the spike protein: a new hope for COVID-19 antibody therapy,” Signal Transduct Target Therapy, vol. 5, no. 208, 2020. Available: https://doi.org/10.1038/s41392-020-00320-6.
[13] C. Zhang et al., “Development and structural basis of a two-MAb cocktail for treating SARS-CoV-2 infections,” Nature Communications., vol. 12, no. 264, 2021. Available: https://doi.org/10.1038/s41467-020-20465-w.
[14] J. C. Santos and G. A. Passos, “The high infectivity of SARS-CoV-2 B.1.1.7 is associated with increased interaction force between Spike-ACE2 caused by the viral N501Y mutation,” bioRxiv, p. 2020.12.29.424708, 2021. Available: https://doi.org/10.1101/2020.12.29.424708.
[15] A. J. Greaney et al., “Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition,” Cell Host & Microbe., vol. 29, no. 1, pp. 44-57, 2020. Available: https://doi.org/10.1016/j.chom.2020.11.007.
[16] J. Lan et al., “Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor,” Nature, vol. 581, no. 7807, pp. 215-220, 2020. Available: https://doi.org/10.1038/s41586-020-2180-5.
[17] P. Wang et al., ‘Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7’, bioRxiv, p. 2021.01.25.428137, 2021. Available: https://doi.org/10.1038/s41586-021-03398-2.
[18] D. O. Ricke, “Two Different Antibody-Dependent Enhancement (ADE) Risks for SARS-CoV-2 Antibodies,” Front. Immunol., vol. 12, 2021. Available: https://doi.org/10.3389/fimmu.2021.640093.
[19] L. B. Rodda et al., “Functional SARS-CoV-2-Specific Immune Memory Persists after Mild COVID-19," Cell, vol. 184, no. 1, pp. 196-183, 2021. Available: https://doi.org/10.1016/j.cell.2020.11.029.
[20] R.-M. Lu et al., “Development of therapeutic antibodies for the treatment of diseases,” Journal of Biomedical Sciences, vol. 27, no. 1, 2020. Available: https://doi.org/10.1186/s12929-019-0592-z.
[21] Department of Business, Enterprise and Innovation, ”Focus on Biopharmachem,” 2018. [Online]. Available: https://enterprise.gov.ie/en/Publications/Publication-files/Focus-on-Biopharmachem-2020.pdf. [Accessed February 2021].
[22] A. Baum et al., “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies,” Science, vol. 369, no. 6506, pp. 1014-1018, 2020. Available: https://doi.org/10.1126/science.abd0831.
[23] E. C. Sabino et al., “Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence,” The Lancet, vol. 397, no. 10273, pp. 452-455, 2021. Available: https://doi.org/10.1016/S0140-6736(21)00183-5.
[24] I. Choi et al., “Machine Learning Methods Enable Predictive Modeling of Antibody Feature: Function Relationships in RV144 Vaccinees,” PLoS Computational Biology, vol. 11, no. 4, p. e1004185, 2015. Available: https://doi.org/10.1371/journal.pcbi.1004185.
[25] L. DeFrancesco, “COVID-19 antibodies on trial,” Nature Biotechnology, vol. 38, no. 11, pp. 1242-1252, 2020. Available: https://doi.org/10.1038/s41587-020-0732-8.
[26] H. Yao et al., “Rational development of a human antibody cocktail that deploys multiple functions to confer Pan-SARS-CoVs protection,” Cell Research, vol. 31, no. 1, pp. 25–36, 2021. Available: https://doi.org/10.1038/s41422-020-00444-y.
[27] L. Hanke et al., “An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction,” Nature Communications, vol. 11, no. 1, pp. 4420, 2020. Available: https://doi.org/10.1038/s41467-020-18174-5.
[28] H. Ma et al., “Potent Neutralization of SARS-CoV-2 by Hetero-Bivalent Alpaca Nanobodies Targeting the Spike Receptor-Binding Domain,” Journal of Virology , vol. 95, no.10, 2021. Available: https://doi.org/10.1128/JVI.02438-20.
[29] A. Rambaut et al., “A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology,” Nature Microbiology, vol. 5, no. 11, pp. 1403-1407, 2020. Available: https://doi.org/10.1038/s41564-020-0770-5.
[30] H. M. Berman et al., “The Protein Data Bank,” Nucleic Acids Research, vol. 28, no. 1, pp. 235–242, 2000. Available: https://doi.org/10.1093/nar/28.1.235.
[31] M. Gouy, S. Guindon, and O. Gascuel, “SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building,” Molecular Biology And Evolution, vol. 27, no. 2, pp. 221–224, 2010. Available: https://doi.org/10.1093/molbev/msp259.
[32] E. F. Pettersen et al., “UCSF Chimera--A visualization system for exploratory research and analysis,” Journal of Computational Chemistry, vol. 25, no. 13, pp. 1605–1612, 2004. Available: https://doi.org/10.1002/jcc.20084.
[33] T. D. Goddard et al., “UCSF ChimeraX: Meeting modern challenges in visualization and analysis,” Protein Science: A Publication Of The Protein Society, vol. 27, no. 1, pp. 14–25, 2018. Available: https://doi.org/10.1002/pro.3235.
[34] R. E. Chen et al., “Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies,” Nature Medicine, vol. 27, no. 4, pp. 717–726, 2021. Available: https://doi.org/10.1038/s41591-021-01294-w.
[35] W. Dejnirattisai et al., “Antibody evasion by the P.1 strain of SARS-CoV-2," Cell, vol. 184, no. 11, pp. 2939-2954, 2021. Available: https://doi.org/10.1016/j.cell.2021.03.055.
[36] W. Dejnirattisai et al., “The antigenic anatomy of SARS-CoV-2 receptor binding domain,” Cell, vol. 184, no. 8, pp. 2183-2200, 2021. Available: https://doi.org/10.1016/j.cell.2021.02.032.
[37] G. Lawton, “Are booster shots coming?,” New Scientist, vol. 250, no. 3334, pp. 8–9, 2021. Available: https://doi.org/10.1016/S0262-4079(21)00808-3.
[38] M. C. Rahalkar and R. A. Bahulikar, “Lethal Pneumonia Cases in Mojiang Miners (2012) and the Mineshaft Could Provide Important Clues to the Origin of SARS-CoV-2," Frontiers In Public Health, vol. 8, 2020. Available: https://doi.org/10.3389/fpubh.2020.581569.
Appendices
Appendices are available in the PDF version of this article.
About This Article
Peer review information: Primary Handling Editor: Kerena Norris. Managing Editor: Anne-Rosa Bilal. Youth STEM Matters thanks Elizabeth Bourn, Dr Luiza Mendonca and Prof Andrew Ward for their contribution to the peer review of this work.
Received: 17 June 2021
Accepted: 2 October 2021
Published: 29 March 2022